Things that we do
Synthetic Methodology using organoboranes and organometallics with applications to bio-organic chemistry.
Cole Research Group – Fall 2013
One of our first major projects was the development of transmetallation reaction of alkyl and alkenyl groups from biscyclopentdienlyzirconcene compounds to boron halides. Examples of these transmetallation reactions are summarized below:
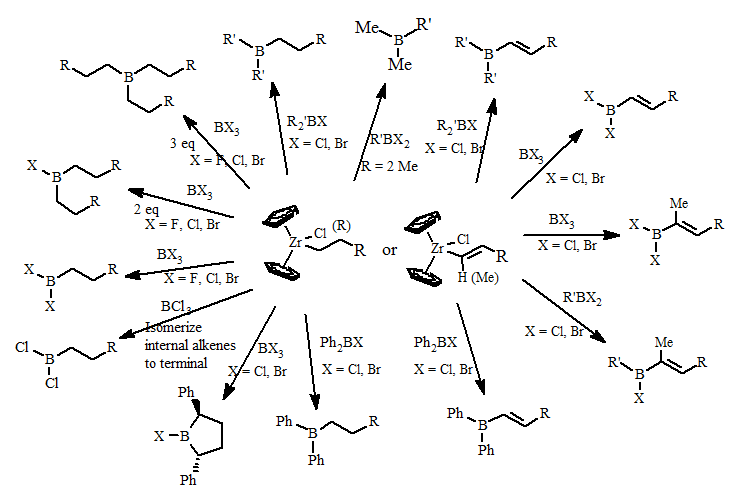
These reactions form organoboranes in higher regiopurity than can be formed using classical organoborane reactions. This opens up the possibility of making new types of organoboranes previously unavailable of investigation and applications to synthetic methodology.
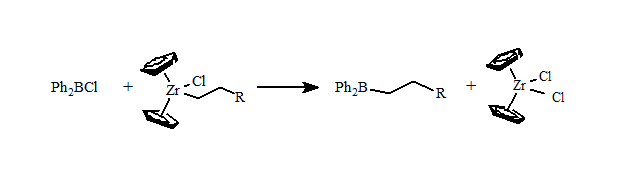
One of my Ph.D. students, Frank Mizori, developed the first general route to alkyldiphenyl- and alkenyldiphenylboranes using a zirconium to boron transmetallation reaction.
These mixed boranes undergo fast 1,4-additions to ?,?-unsaturated aldehydes and ketones in high yields. They also can be used for the reductive alkylation of p-benzoquinone to form the alkylhydroquinones in good yields.
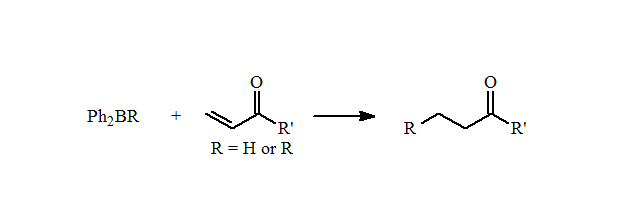
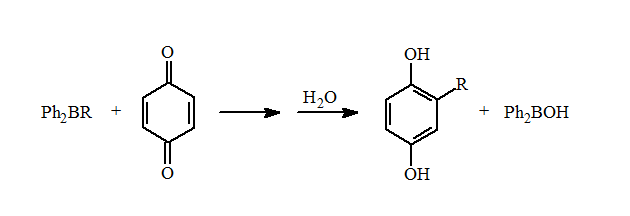
Unfortunately, there is only a limited number of alkyl types can be incorporated into these diphenylalkylboranes using this reaction. Importantly, these reactions demonstrated that mixed organoboranes gave good yields in the reductive alkylation of quinones and 1,4-additions.
Reductive Alkylation of Quinones
The reductive alkylation of p-benzoquinone is one of the few unsuccessful organoborane reactions. This was also the first carbon-carbon bond forming reaction discovered after the development of the hydroboration reaction in 1956. This reaction never lived up to its potential nor did the 1,4-addition to ?,?-unsaturated aldehydes and ketones. These reactions had a number of characteristics that make it different from many of the other types of organoborane reactions.
Limitations in the reductive alkylation reaction:
- Only one of the three groups is transferred to product with a loss of 2 alkyl groups.
- These reactions have an unusual migratory aptitude, 3° > 2° > 1° and phenyl does not migrate. Most organoborane reactions show an inverse migratory aptitude, with the less substituted alkyl groups migrating faster than more substituted groups.
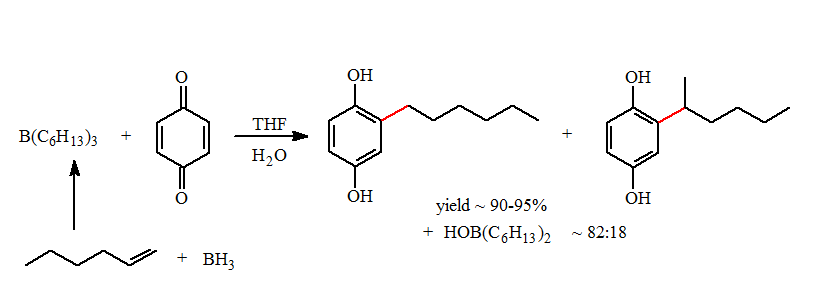
- Borane, (BH3.THF, BH3.SMe2 and etc) are low regioselective borane reagents in the hydroboration reaction. In the case of 1-hexene this is approximately 95:5. The more selective hydroborating reagents; such as, Cy2BH, 9-BBN-H, Sia2BH cannot be used since the secondary auxiliary groups will migrate faster than a desired primary alkyl group and migrate competitively with a secondary alkyl group. There were no solutions to preparing mixed boranes containing low migratory aptitudes groups until recently.
- As a consequence of the migratory aptitude and the formation of regioisomers formed with BH3, a mixture of hydroquinone products were formed in this reaction. In the case of the trihexylborane the 1-hexyl- to 2-hexylhydroquinone was formed in an approximate ratio of 82:18. Essentially all of the 2-hexyl groups are transferred in the reductive alkylation. Mixtures of isomeric products are difficult to separate and this lowers of the desired product yield.
- Finally, the borinic ester, R2BOH, formed on hydrolysis is air-sensitive. On exposure to oxygen, this results in the formation of radicals. Hydroquinones are known radical traps and the alkylhydroquinones are converted into a dark colored polymeric tarry material. Also, due to size of the alkyl groups, the borinic acids are difficult to separate from the hydroquinone product.
For these reasons, the reductive alkylations of quinones have received little attention or application.
We began our investigations when Frank Mizori (Ph.D. 2004) demonstrated that Ph2BR formed the alkylated hydroquine product. But the types of alkyl groups that can be prepared by these zirconium transmetallations are limited.
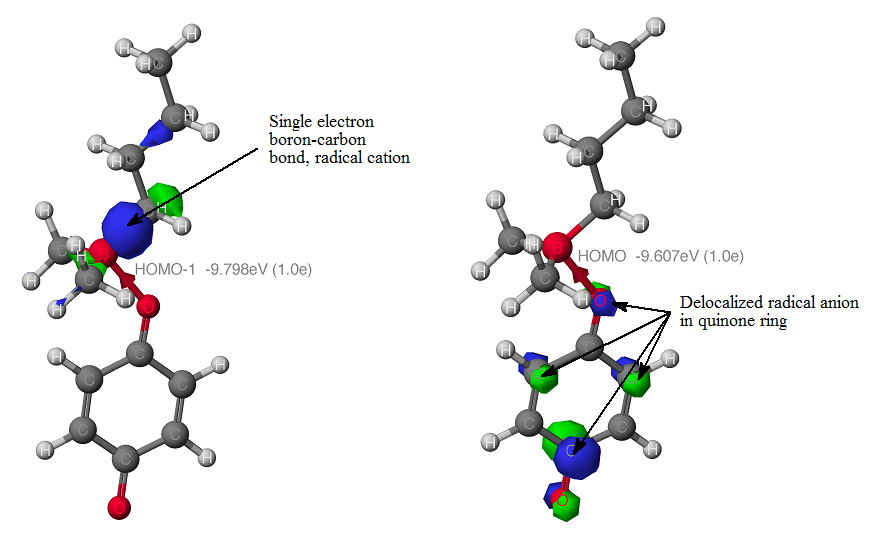
A review of the organoborane reductive alkylation literature suggests a free radical intermediate. The migratory aptitude follows free radical stability of the alkyl group. Therefore, methyl groups, like the phenyl group, should be good auxiliary groups for primary and secondary alkyl groups. This was supported by our calculations in which a single electron is transferred from the more substituted R-B bond (HOMO orbital) into the quinone ring.
The preparation of alkylmethylboranes has long been known to give low yields and form mixtures of redistribution products. This facile redistribution has hindered the development of these mixed alkylmethylboranes.
We began our investigation of this chemistry acknowledging there were a number of challenges that needed to be solved.
1. Preliminary attempts to form the selective hydroborating reagent, HBCl2.L by redistribution failed to cleanly form this reagent. Recently, a number of research groups have reported similar results. Despite earlier literature, this method does not form the dichloroborane with selectivity greater than 75 mol% HBCl2.L or RBCl2 after hydroboration.
Mattesons in situ preparation of HBCl2 from BCl3/Et3SiH and hydroboration gives excellent formation of the desired RBCl2 = 98 mol% purity. This is an excellent and selective route to the alkyldichloroboranes.
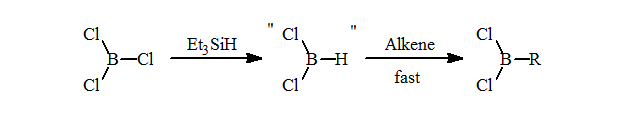
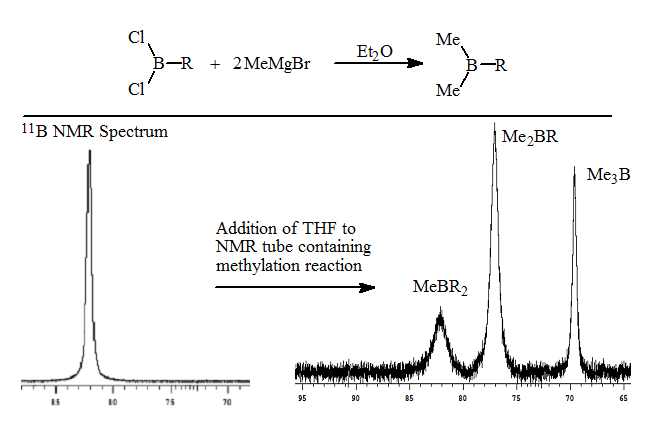
2. The methylation of the RBCl2 with MeMgBr proceeds smoothly to form the trialkylborane as seen in the 11B NMR spectrum.
Until recently, redistribution was detected using either carboxylation/oxidation, DCME/base/oxidation or cyanidation/oxidation reactions followed by capillary GC analysis of the tertiary alcohol products. These methods require 1-2 days and are somewhat tedious.
The facile redistribution of methylalkylboranes has been a long-standing problem that has hindered the investigation and development of these boranes.
3. Addition of a weak coordinating ligand such as THF results in the separation of the methylalkylboranes based on the number of methyl groups present. The trimethylborane resonance was shifted furthest upfield, while methyldialkylborane shifted least amount on complexation. The ratios of the methylalkylboranes were 20:60:20. The 11B NMR analysis takes approximately 10-15 minutes. These results are described in a manuscript in preparation.
This rapid analysis allowed us to readily explore the factors that resulted in redistribution. The addition of the alkyldichloroborane to a THF solution of the methylmagnesium bromide resulted in no detectable redistribution as seen in the following spectrum.
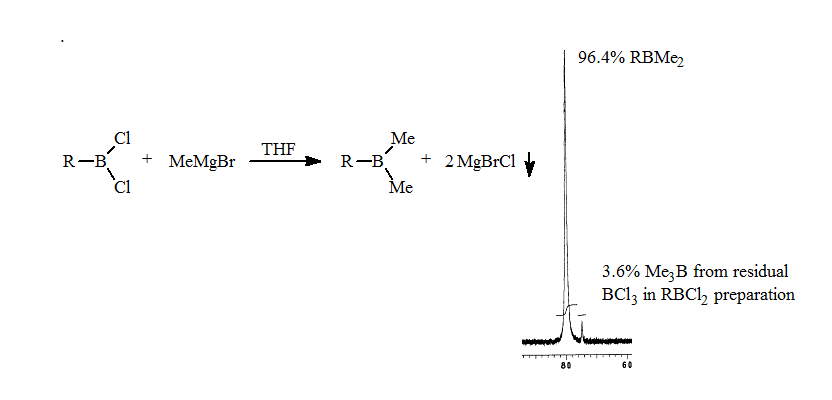
This development gave the first general route to pure alkyldimethylborane.
4. However, the reductive alkylation of p-benzoquinone with the alkyldimethylborane prepared above resulted in an approximately 50% yield of the expected alkylhydroquinone. A careful analysis showed the missing mass to be the simple reduced hydroquinone.
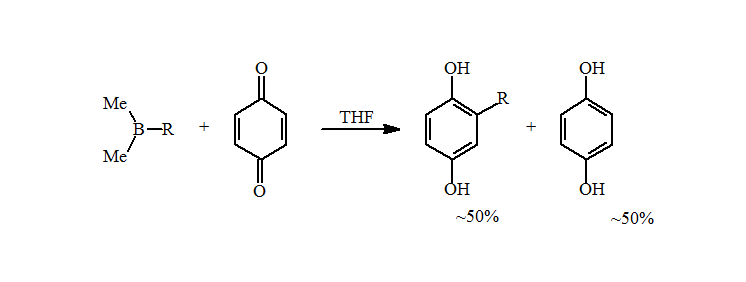
5. This problem was not seen when the trialkylboranes were prepared by hydroboration. The presence of metals, such as magnesium or lithium, catalyzes the reduction of the quinone to the hydroquinone with a loss of an alkyl group. A simple water wash is sufficient to remove the metals and increase the amounts of the alkylhydroquinone to near quantitative isolated yields for primary alkyl groups.
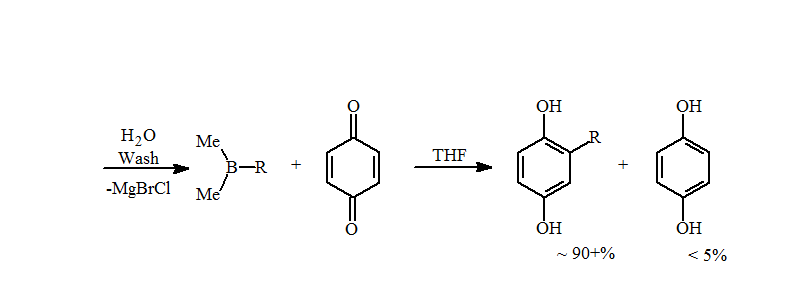
The reaction is very rapid, as seen in the loss of the quinone yellow color within a second of addition. After approximately 15 min, the volatiles are removed under reduced pressure, which also removes the Me2BOH, avoiding formation of radicals and loss of hydroquinone product. The alkylhydroquinone products are isolated as an off-white solid in high yields.
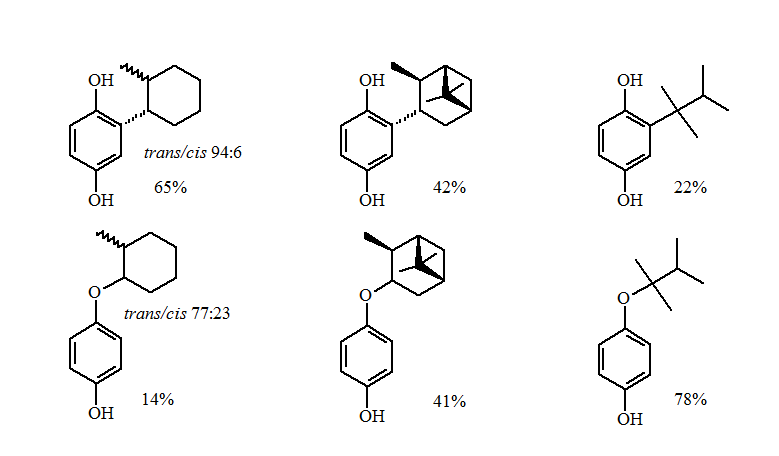
The reductive alkylations of primary and non-sterically hindered secondary groups gave excellent yields of the alkylhydroquinone.
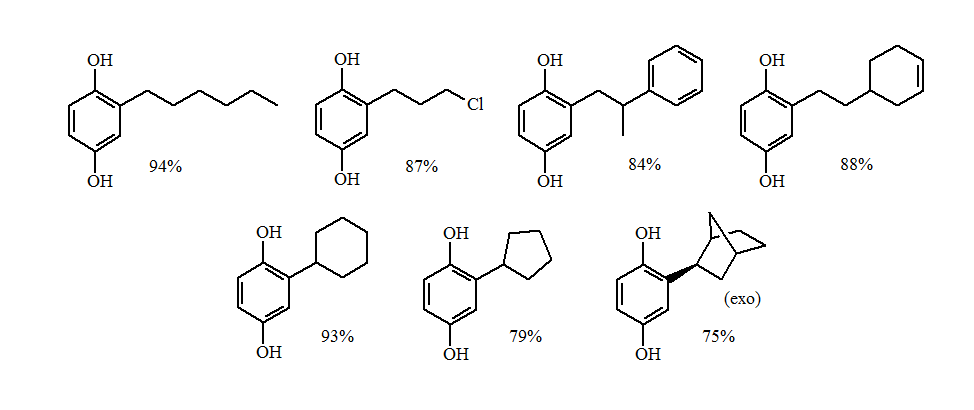
The more sterically demanding secondary and tertiary groups gave a loss of stereochemistry and formation of the O-alkylation products. The greater the steric demands the larger the amounts of O-alkylation.
While these reactions represent the most successful organoborane route to alkylhydroquinones via reductive alkylation, the alkyl groups are largely limited to saturated alkyl groups and only a few types of functional groups are considered to be compatible with boranes.