Yuezhi Mao
Assistant Professor, Physical and Computational Chemistry
office: GMCS 213D
phone: 619-594-1617
email: ymao2@sdsu.edu

Mao Group Page
Curriculum Vitae
- B.Sc., Materials Chemistry, Peking University, 2012
- Ph.D., Chemistry, U.C. Berkeley, 2017
- Postdoc, Stanford University, 2018-2022
- Assistant Professor, San Diego State University, 2022-present
Research Interests
The research of the Mao group centers on the development and application of theoretical and computational methods to elucidate the fundamental mechanisms involved in chemical problems ranging from molecular catalysis to photochemical and electron transfer processes in the condensed phase. These problems present forefront challenges to the current theoretical and computational chemistry research, since they require a combination of tools to accurately and efficiently capture the ground and excited state potential energy surfaces (PESs), to simulate dynamical processes in complex environments of a wide range of timescales, and to bridge the computational and simulation results with chemical intuitions and experimental observations. Our lab is aiming to address these challenges by developing electronic structure and multiscale modeling methods, and by embracing new emerging tools such as machine learning (ML). By applying these new methods to the cutting-edge chemical problems, we will be able to extract new design principles for molecular catalysts and uncover intriguing excited-state processes pertinent to the design of functional molecules such as those for light-emitting devices and bio-imaging sensors. Specifically, our group will explore the following research directions:
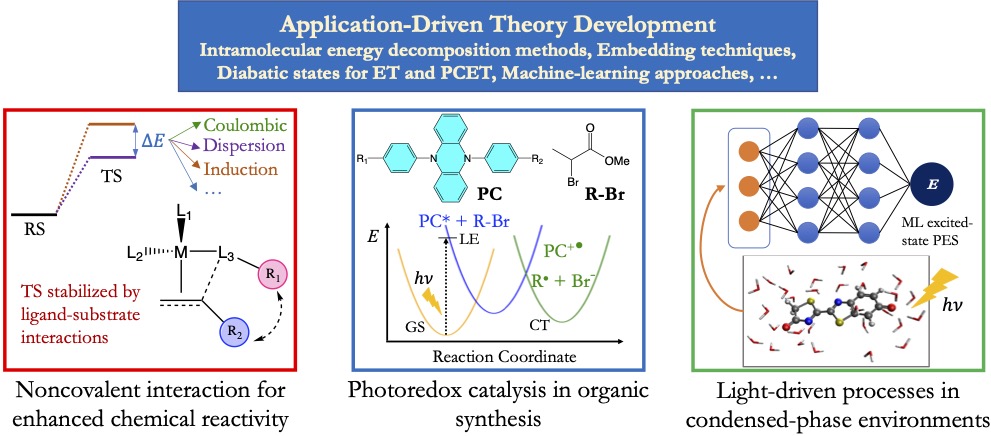
Harnessing noncovalent interactions in catalyst design
Noncovalent interactions (NCIs), including components such as electrostatics, steric repulsion, and dispersion, play an important role in molecular, biochemical, and interfacial catalysis. It has thus been a common practice for chemists to make chemical modifications to tune these NCIs to achieve improved chemical reactivity and selectivity. However, these modifications do not always work as one would expect based on one’s chemical intuition, and consequently catalyst design still involves a lot of trial and error. This mainly results from the competition between different components of NCI as well as their sensitivity to chemical positioning and the surrounding environments.
Our lab will develop both physically motivated and data-driven approaches to help understand the role of NCIs in molecular catalysis and to facilitate the modular design of new catalysts leveraging these interactions. We will introduce a new interaction partitioning scheme that can precisely identify how each specific part of a catalyst contributes to the stabilization of the transition states. This will be able to tell us which part of the molecular catalyst should be modified and how this modification might look like. We will then leverage ML to accelerate the screening of candidate catalysts with the prediction of reaction barriers using reactant state properties, including those obtained from the decomposition of substrate-catalyst interactions. In addition, to facilitate the experimental verification of catalyst design leveraging NCIs, we will build quantitative or semi-quantitative maps between spectroscopic shifts and physical descriptors of NCIs that can be obtained from calculations.
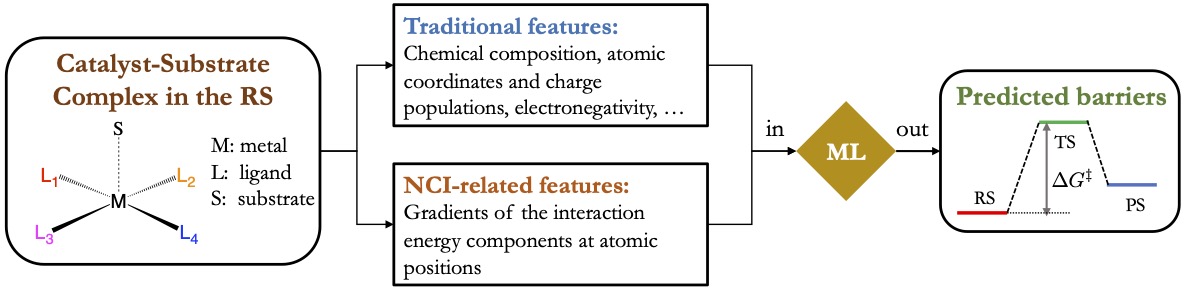
Elucidating mechanisms of organic photoredox catalysis
Photoredox catalysis is a powerful technique that uses photoexcitation to transform weakly reactive closed-shell species into highly reactive radicals. While traditionally people have been employing heavy transition metal (TM) complexes, such as those of Ir and Ru, to catalyze photoredox reactions, recently several promising catalysts based purely on organic molecules have been proposed. These organic photoredox catalysts (PCs) are potentially more suitable for scalable productions because of their lower cost and environment toxicity. Unlike TM-based catalysts that typically have long-lived triplet states, the organic PCs use their much shorter-lived S1 and/or T1 states as the redox-active species. Therefore, for the design of organic PCs it is important to know how the rate of the electron transfer (ET) or proton-coupled electron transfer (PCET) process compares to those of the competing excited-state decay processes.
Our group will develop a computational framework based on electronic diabatic states to predict the rates of excited-state ET/PCET and those competing processes for the design of purely organic PCs. Those diabatic states localize electronic excitations as well as the transferred electron/proton on specific sites such that they naturally correspond to the initial and final states of an ET/PCET process. Our research will focus on the development of specialized electronic structure methods to construct diabatic states involved in excited-state ET/PCET processes, especially for those short-lived excited states of varying spin-multiplicities, as well as methods to evaluate the couplings between these diabatic states. These developments will allow us to predict the rates of the redox and the competing excited-state decay processes in silico for organic PCs using rate theories or quantum dynamics methods. Doing these calculations for a large pool of PC candidates, we are aiming to establish the relationship between the reactivity of organic PCs with their intrinsic properties as well as the important environment factors.
Decoding the role of condensed-phase environments in light-driven chemical processes
Understanding the impact of complex environments on light-driven chemical processes is of great importance to applications such as solar energy conversion, photoluminescent materials, bio-imaging, and human vision. Recent advances in time-resolved and multidimensional spectroscopy have provided new powerful tools to probe excited-state dynamics in various condensed-phase environments, which have motivated the development of theoretical approaches to unravel the chemical mechanisms underlying those measured spectra. For that purpose, on-the-fly dynamics simulations stand for a powerful tool since they can provide atomistic details for the photochemical processes. However, treating the environment at a sufficient level of accuracy entails high computational costs, especially in on-the-fly dynamics simulations where the energies and forces need to be calculated at each time step.
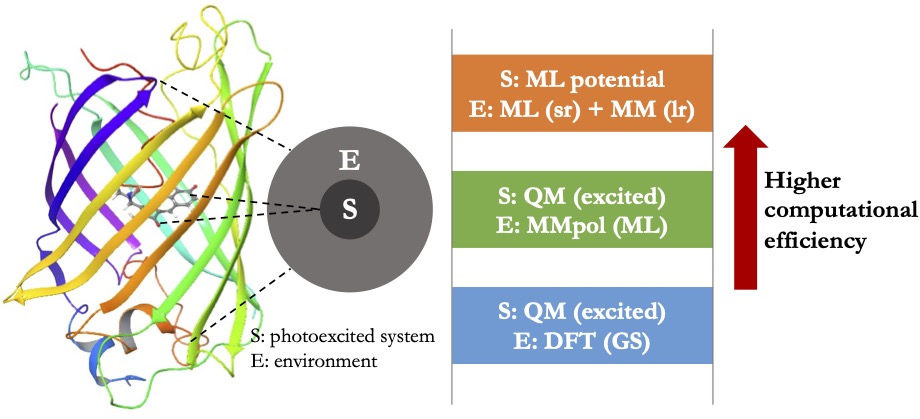
Our goal is to construct accurate PESs that can be utilized for efficient on-the-fly dynamics simulations of excited-state processes in condensed-phase environments. Using these simulations, we aim to elucidate the effects of the environment on photochemical processes and provide microscopic details for the interpretation of time-resolved spectroscopy. To achieve this, we will develop a hierarchy of multiscale modeling methods ranging from quantum mechanical (QM) embedding theory to ML-assisted PESs (Figure 2). These developments will offer a powerful toolbox for modeling excited-state systems in complex environments at different levels of accuracy and computational cost, facilitating our understanding of how important light-driven chemical processes can be modulated and controlled by modifying the chemical environment.
Students who join the Mao lab are expected to acquire the following skills: (i) usage of computational chemistry software packages for electronic structure calculations (Q-Chem, ORCA, CP2K, etc.) and molecular dynamics simulations (e.g., GROMACS); (ii) data processing, analysis, and visualization using Python or other tools; (iii) construction of machine-learning models using Python and specialized featurization schemes for chemical systems; (iv) coding in the Q-Chem program (mainly in C++) for the development and improvement of electronic structure methods. Our group is looking for new members with a background in chemistry, physics, computer science, or other related areas who are interested in developing new computational chemistry tools and/or applying these tools to solve problems in chemsitry. For additional information, please contact the PI at ymao2@sdsu.edu.
Selected Publications
- C. Zheng, Y. Mao, J. Kozuch, A. O. Atsango, Z. Ji, T. E. Markland, and S. G. Boxer, “A two-directional vibrational probe reveals different electric field orientations in solution and an enzyme active site,” Nat. Chem., in press. doi: 10.1038/s41557-022-00937-w (as co-first author)
- Y. Mao, M. Loipersberger, P. R. Horn, A. Das, O. Demerdash, D. S. Levine, S. P. Veccham, T. Head-Gordon, and M. Head-Gordon, “From intermolecular interaction energies and observable shifts to component contributions and back again: A tale of variational energy decomposition analysis,” Annu. Rev. Phys. Chem., 2021, 72, 641-666. doi: 10.1146/annurev-physchem-090419-115149
- Y. Mao, M. Loipersberger, K. J. Kron, J. S. Derrick, C. J. Chang, S. M. Sharada, and M. Head-Gordon, “Consistent Inclusion of Continuum Solvation in Energy Decomposition Analysis: Theory and Application to Molecular CO2 Reduction Catalysts,” Chem. Sci., 2021, 12, 1398-1414. doi: 10.1039/D0SC05327A
- E. Epivanovsky, A. T. B. Gilbert, X. Feng, J. Lee, Y. Mao, and N. Mardirossian et al., “Software for the frontiers of quantum chemistry: An overview of developments in the Q-Chem 5 package,” J. Chem. Phys., 2021, 155, 084801. doi: 10.1063/5.0055522 (as a major contributor to the Q-Chem software)
- Y. Mao, A. Montoya-Castillo, and T. E. Markland, “Excited state diabatization on the cheap using DFT: Photoinduced electron and hole transfer,” J. Chem. Phys., 2020, 153, 244111. doi: 10.1063/5.0035593
- Y. Mao, D. S. Levine, M. Loipersberger, P. R. Horn, and M. Head-Gordon, “Probing Radical-Molecule Interactions with a Second Generation Energy Decomposition Analysis of DFT Calculations Using Absolutely Localized Molecular Orbitals,” Phys. Chem. Chem. Phys. (perspective), 2020, 22, 12867-12885. doi: 10.1039/D0CP01933J
- Y. Mao, A. Montoya-Castillo, and T. E. Markland, “Accurate and efficient DFT-based diabatization for hole and electron transfer using absolutely localized molecular orbitals,” J. Chem. Phys., 2019, 151, 164114. doi: 10.1063/1.5125275
- Y. Mao and M. Head-Gordon, “Probing Blue-Shifting Hydrogen Bonds with Adiabatic Energy Decomposition Analysis,” J. Phys. Chem. Lett., 2019, 10, 3899-3905. doi: 10.1021/acs.jpclett.9b01203
- Y. Mao, M. Head-Gordon, and Y. Shao, “Unraveling Substituent Effects on Frontier Orbitals of Conjugated Molecules Using an Absolutely Localized Molecular Orbital Based Analysis,” Chem. Sci., 2018, 9, 8598-8607. doi: 10.1039/C8SC02990C
- Y. Mao, Q. Ge, Paul R. Horn, and M. Head-Gordon, “On the Computational Characterization of Charge- Transfer Effects in Non-Covalently Bound Molecular Complexes,” J. Chem. Theory Comput., 2018, 14, 2401-2417. doi: 10.1021/acs.jctc.7b01256
- Y. Mao, P. R. Horn, and M. Head-Gordon, “Energy decomposition analysis in an adiabatic picture,” Phys. Chem. Chem. Phys., 2017, 19, 5944-5958. doi: 10.1039/C6CP08039A
- Y. Mao, Y. Shao, J. Dziedzic, C.-K. Skylaris, T. Head-Gordon and M. Head-Gordon, “Performance of the AMOEBA water model in the vicinity of QM solutes: A diagnosis using energy decomposition analysis,” J. Chem. Theory Comput., 2017, 13, 1963-1979. doi: 10.1021/acs.jctc.7b00089
- Y. Mao, P. R. Horn, N. Mardirossian, T. Head-Gordon, C.-K. Skylaris, and M. Head-Gordon, “Approaching the basis set limit for DFT calculations using an environment-adapted minimal basis with perturbation theory: Formulation, proof of concept, and a pilot implementation,” J. Chem. Phys., 2016, 145, 044109. doi: 10.1063/1.4959125
- Y. Mao, O. Demerdash, M. Head-Gordon, and T. Head-Gordon, “Assessing Ion-Water Interactions in the AMOEBA Force Field Using Energy Decomposition Analysis of Electronic Structure Calculations,” J. Chem. Theory Comput., 2016, 12, 5422-5437. doi: 10.1021/acs.jctc.6b00764