11B NMR Chemical Shifts
(Relative to BF3.Et2O)
Cole Research Group, SDSU Department of Chemistry & Biochemistry
Common Boron Chemical Shifts
The following collection of 11B NMR chemical shifts is a selected compilation of representative organoborane and boron compounds that we believe would be of interest to the organoborane community and to my research group at San Diego State University. There are a variety of types of borane compounds that are absent from this compilation, including the carboranes, metal complexes, etc. While we have made a reasonable effort to ensure the accuracy of this data, errors are probably inevitable. Corrections and additions of important types of boranes are most welcome. Please email along with your additions, corrections or comments here: tcole@sciences.sdsu.edu.
- Index
- General Comments on the Boron Chemical Shifts
- Summary of B-11 Chemical Shifts
- General Literature References
- Catalog
-
General Comments on the Boron Chemical Shifts
Coordination
The addition of a ligand or base to the empty p-orbital on boron results in an upfield shift as compared to the tricoordinate borane. This effect is also seen with coordinating solvents such as THF. The chemical shift is dependent on the strength of the coordination complex, with the stronger complexes shifted to higher field. Borane dimers are also found upfield from the free monomer and may show a more complex coupled spectra due to the presence of both terminal and bridging hydrogens coupling to the boron. The addition complex with solvent or a basic ligand is generally found higher field than the dimer.
Tricoordinate Boranes Containing Hydrogen
The coupled 11B NMR spectra generally shows a B-H coupling, unless there is a fast exchange of the hydrogens on the NMR time scale. Since the spin of hydrogen is 1/2, the number of peaks in the multiplet is one greater than the number of hydrogens attached to the boron. The magnitude of the B-H splitting increases with additional ligands and greater electronegativity of the ligand. The trivalent boranes tend to be dimeric, especially in non-coordinating solvents in the absence of sterically large groups. In a number of cases one may observe both the monomeric borane (non-coordinated to the solvent or any another weak base such as dimethyl sulfide) and the dimeric borane. The chemical shifts of the trivalent organoboranes having a B-H bond are more variable than that of the trialkylboranes, and are dependent on the structure of the organic groups present.
Trialkylboranes
The trialkylboranes are found in a narrow low field region, 83-93 ppm. These chemical shifts are largely independent of the structure of the alkyl group. The exception are for the tertiary groups that shift the resonance ~ 3 ppm upfield. Cyclic boranes show a marked dependence on the size of the ring. Six membered rings have approximately the same chemical shifts as the acyclic compounds. The borolane compounds, five member rings, are shifted to lower field by ~ 6 ppm. The effect of the ring size is observed for all cyclic boranes independent of the substitutents present.
Substitution of Alkyl Groups
Substitution at the a -carbon, R2B-CH2-X, with X = N3, OH, NH2, Cl, Br, I, PMe3, AsMe3, SMe, BR2, SiR3, Ph, vinyl results in an upfield shift of the borane. This effect may be attributed to inter- or intramolecular association of the lone electron pair with the empty pi-orbital on the boron or a neighboring group anisotropic effect. The degree of upfield shift is variable, Cl is relatively small ~ +2.9 ppm while I is much larger ~ 15.4 ppm. Silyl and boryl groups shift upfield ~ 4-8 ppm. Similar explanation is made for phenyl and vinyl groups, ~ 0 - 6 ppm.
Boron compounds that are directly bonded to sp2 or sp carbons are shifted to higher field due to a pi-interaction between the two p-orbitals on the adjacent carbon and boron atoms. Steric effects are also very important, especially in the case of weaker pi-donors. The effects of acetylenic groups is larger than that of the alkenyl groups, probably due to anisotropic effects as opposed to pi-orbital interactions.
The presence of an OR' or OH group bonded to the boron results in 11B resonance at higher fields as compared to the corresponding alkylborane. The OR' and OH groups are strong pi-donors. In general, the structures of the alkyoxy groups do not effect the boron chemical shifts for oxygen containing tricoordinate boranes. The exceptions to this are the tert -butyl, phenoxy and Me3Si groups which give slightly higher upfield shift of ~ 2-4 ppm.
Boroxines
The cyclic anhydrides of the boronic acids are the boroxines, (RBO)3. These compounds are found at slightly lower field, ~ 33 ppm, that the corresponding boronic esters or acids, ~ 30 ppm. Aryl and vinyl boroxines, are shifted upfield approximately 2-4 ppm from the alkyl derivatives.
Borohydrides
The parent borohydrides, BH4-, is a tetrahedral compound in which all hydrogens are magnetically equivalent. A quintet is observed in a 1:4:6:4:1 ratio. While the 1H NMR spectra shows a 1:1:1:1 resonance. The borohydrides may be ionic or covalent in nature and this may result in a more complex spectra depending on the interactions of the cation with the borohydride hydrogens. In most cases, the exchange is fast and only one type of hydrogen is observed. The boron resonances are shifted to higher field, -26 to -45 ppm, more than most all other boron species. The solvent has a significant effect on the chemical shift due to solvation of the cation.
Replacement of a hydrogen with an alkyl group shifts the resonances downfield. The 11B NMR signal of these substituted alkylborohydrides, R4-nBHn overlap but the number of hydrogens present can readily be determined from the multiplicity of the proton coupled boron NMR.
Tetraalkylborates
The tetraalkylborates do not follow this pattern. These complexes are observed over a fairly narrow region, -15 to -22 ppm. The replacement with an aryl group shifts these tetraorganylborates downfield in a systemic manner. The tetraphenylborates are seen ca -6.0 ppm and in general all of these borates show a slight dependence on the cation and solvent.
Organylalkoxy Borates
The tetracoordinate alkyl- or arylalkoxyborates are more difficult to generalize in their chemical shifts. Relatively few of these types of compounds have been reported. The majority of these compounds are observed between +12 and -8 ppm. As before, the observed chemical shift is dependent on the cation, concentration and solvent.
Summary of B-11 Chemical Shifts
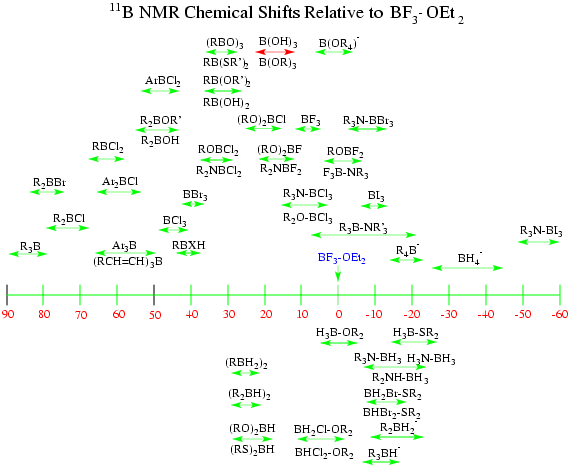
General Literature References
1. R. Schaeffer, "NMR of Boron Hydrides and Related Compounds," Prog. Boron Chem., 1, 417 (1964).
2. G. R. Eaton, "NMR of Boron Compounds," J. Chem. Ed., 46, 547 (1969).
3. G. R. Eaton and W. N. Lipscomb, "NMR Studies of Boron Hydrides and Related Compounds," Benjamin, NY, 1969.
4. W. G. Henderson and E. F. Mooney, "Boron-11 NMR Spectroscopy," Ann. Rev. NMR Spectrosc., 2, 219 (1969).
5. H. Beall and C. H. Bushweller, "Dynamic Processes in Boranes, Borane Complexes, Carboranes, and Related Compounds," Chem. Rev., 73, 465 (1973).
6. W. L. Smith, "Boron-11 NMR," J. Chem. Ed., 54, 469 (1977).
7. H. Nöth and B. Wrackmeyer, "NMR Spectroscopy of Boron Compounds," NMR Basic Prin. Prog., 14, 1 (1978).
8. L. J. Todd and A. R. Siedle, "NMR Studies of Boranes, Carboranes and Heteroatom Boranes," Prog. Nucl. Magn. Reson. Spectrosc., 13, 87 (1979).
9. B. Wrackmeyer, "Carbon-13 NMR Spectroscopy of Boron Compounds," Prog. Nucl. Magn. Reson. Spectrosc.,12, 227 (1979).
10. A. R. Siedle, "Boron-11 NMR Spectroscopy,"Annu. Rep. NMR Spectrosc., 12, 177 (1982).
11. R. G. Kidd, "Boron-11," in "NMR of Newly Accessible Nuclei," Vol. 2, P. Laszlo, Ed., Academic Press, NY, 1983, Ch. 3.
12. J. D. Kennedy, "Boron," in "Multinuclear NMR," J. Mason, Ed., Plenum Press, NY, 1987, Ch. 8.
13. A. R. Siedle, "Boron-11 NMR Spectroscopy," Annu. Rep. NMR Spectrosc., 20, 205 (1988).
14. B. Wrackmeyer, "NMR Spectroscopy of Boron Compounds Containing Two-, Three- and Four-Coordinate Boron," Annu. Rep. NMR Spectrosc., 20, 61 (1988).
15. S. Hermanek, "11B NMR Spectra of Boranes, Main-Group Heteroboranes, and Substituted Derivatives. Factors Influencing Chemical Shifts of Skeletal Atoms," Chem. Rev., 92, 325 (1992).
Catalog
- BX3 and BX3.L
- HBX2 and HBX2.L
- H2BX and H2BX.L
- BH3 and BH3.L
- MBX4
- MBH4
- MBY4
- MBYH3
- MBY3H
- R3B
- R3B.L
- R2BH
- R2BH.L
- R2BX
- R2BX.L
- R2BO-
- R2BO.L
- R2BN=
- RBH2
- RBH2.L
- RBX2
- RBX2.L
- RB(O-)2
- RB(O-)2•L
- RB(N=)2
- RBYH
- RBYH•L
- RBYZ
- BY3
- BY3•L
- BHY2
- Y2BH•L
- YBH2
- YBH2•L
- Y2BZ
- Y2BZ•L
- BWYZ
- M+ BR4-
- M+ BR3L-
- M+ BR2L2-
- M+ R3BH-
- M+ R2BH2-
- M+ RBH3-
- Miscellaneous